Single Cell
The sequencing of the genome, transcriptome and epigenome of individualised cells has revolutionised many areas of biological research (embryonic development, microbiology, neurobiology, immunology, cancer…). This method enables, for example, to study the lineage of a cell line from stem cell to differentiated cell, or to identify tissue heterogeneity, phenotypic modification according to cell microenvironment, etc. Applied in oncology, it also allows the detection and analysis of tumour cells circulating in the blood at an early stage of the disease and therefore facilitates therapeutic follow-up.
The single cell sequencing process involves several steps (review “Single cell sequencing: a distinct new field” Wang et al Clin and Translational Med 6 :10 (2017) doi : 10.1186/s40169-017-0139-4)
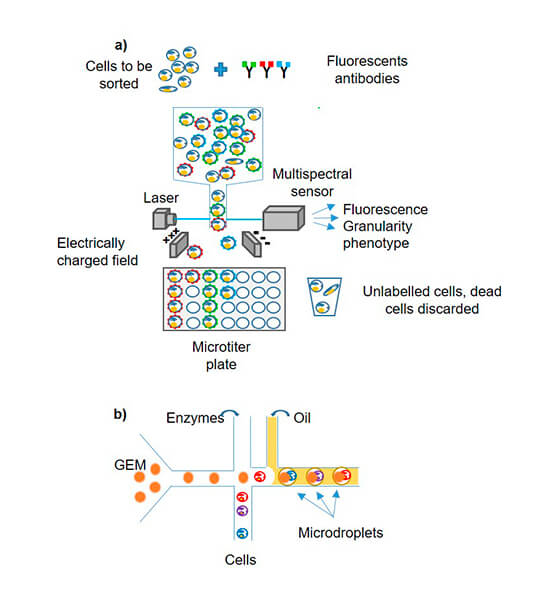
- cell isolation: several cell sorting methods are available depending on the tissue or bio-fluid studied (FACS; LCM etc.) The micro-fluidic tends to supplant other isolation protocols due to its efficiency and production rate, although the FACS allows sorting according to several phenotypic criteria (Fig1).
- amplification: the quantity of nucleotides extracted from a cell (~6 to 10pg) is very limited to build the libraries for sequencing. It is therefore essential to include in the process a total genomic amplification (WGA/WTA: Whole Genome/Trancriptome Ampilfication, MDA: multiple displacement amplification, MALBAC: Multiple annealing and looping based amplification cycles, SMART-seq, etc.)
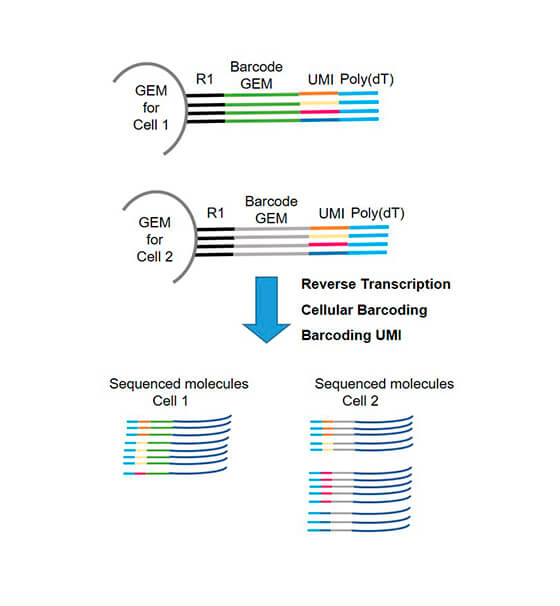
- barcoding: each cell is identified by a unique barcode as well as each amplified cDNA fragment (UMI: Unique Molecular Identifier). The technology using GEM (Gel bead in Emulsion) allows this double barecoding for transcriptome studies (Fig2).